Nonnegative spatial factorization (NSF)
NSF is probabilistic, spatially-aware, interpretable dimension reduction for count data. We illustrate with applications to spatial transcriptomics.
We are not the first to propose spatially-aware dimension reduction. MEFISTO from Britta Velten and Oliver Stegle was a major inspiration. Like PCA and factor analysis, MEFISTO uses real-valued factors, whereas NSF uses nonnegative factors, similar to probabilistic NMF.
The advantage of nonnegative factors is a parts-based representation (face=eyes+nose+mouth). Real-valued models produce holistic representations (“eigenfaces”).
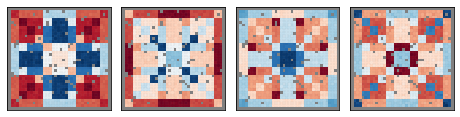

top: MEFISTO holistic representation, bottom: NSF representation, on the same composite samples. red=negative, blue=positive, white=zero.
Nonspatial nonnegative models like probabilistic NMF also learn parts-based representations. The advantage of a spatially-aware model is generalization beyond the training set. NSF uses log-Gaussian processes that produce continuous surfaces across the spatial domain.
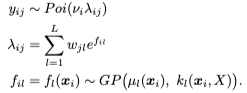
Because some variation may be nonspatial (e.g., blood or immune cells dispersed across a tissue), we combine spatial and nonspatial factors in the NSF hybrid model (NSFH). This model also gives a spatial importance score for each feature (gene) or observation (cell).
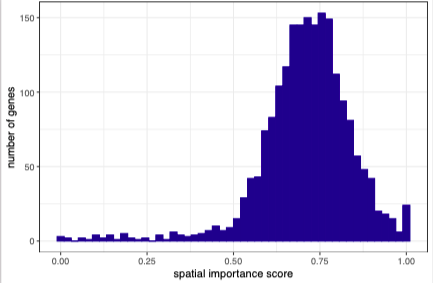
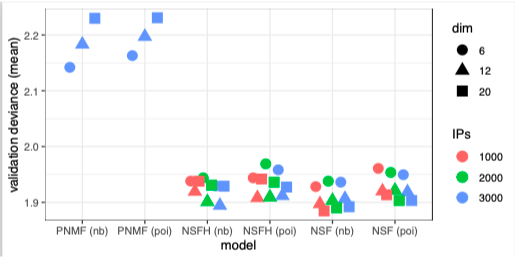
Our spatially-aware NSF and NSFH models exhibit better generalization accuracy (out-of-sample prediction) than nonspatial probabilistic NMF on a benchmarking task.
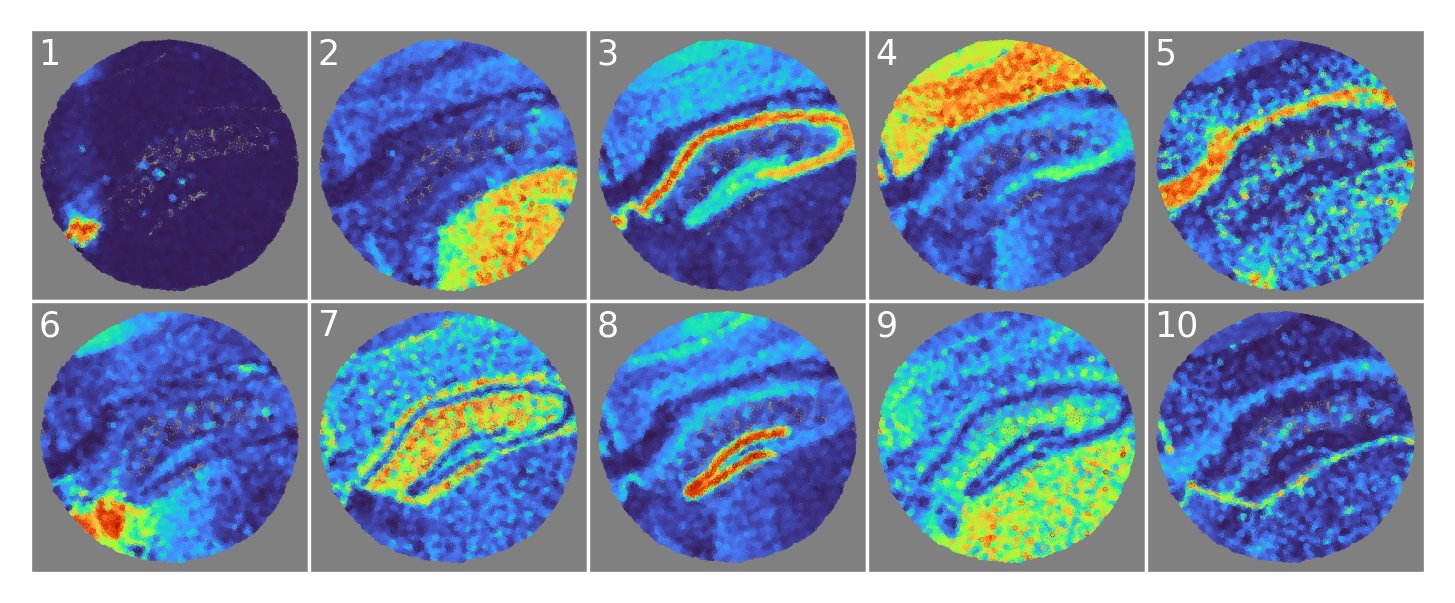
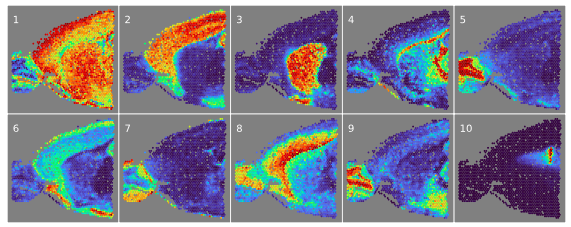
Our NSFH model identified specific brain regions in Slide-seqV2 mouse hippocampus, including choroid plexus (1), thalamus (2), cerebral cortex (4), medial habenula (6), dentate gyrus (8), and even the thin meningeal layer (10).
Here’s what NSFH spatial factors look like on Visium data, again identifying specific brain regions such as basal ganglia (3), corpus callosum (4), and choroid plexus (10).
We use the loadings matrix to find informative features (genes) for each component, and map these to cell types and biological functions. scfind (https://scfind.sanger.ac.uk) from @THJimmyLee @m_hemberg was extremely useful for this task.
We are grateful to the spatial transcriptomics community for making so many fascinating datasets publicly available. @ghartoularos and Dylan Cable of @rafalab answered many of our initial data questions.
We learned a lot from conversations with folks outside of genomics. Spatial and temporal dimension reduction models have a great history in environmental science and diverse applications. @anqiwu_angela and others from @jpillowtime group are doing amazing things in neuroscience.